HHS and NIH Take Steps to Enhance Transparency of Clinical Trial Results

The US Department of Health and Human Services has issued a Notice of Proposed Rulemaking (NPRM), which proposes regulations to implement reporting requirements for clinical trials that are subject to Title VIII of the Food and Drug Administration Amendments Act of 2007 (FDAAA). The proposed rule clarifies requirements to clinical researchers for registering clinical trials and submitting summary trial results information to ClinicalTrials.gov, a publicly accessible database operated by the National Library of Medicine, part of the National Institutes of Health. A major proposed change from current requirements is the expansion of the scope of clinical trials required to submit summary results to include trials of unapproved, unlicensed, and uncleared products.
“Medical advances would not be possible without participants in clinical trials,” said NIH Director Francis S. Collins, MD, PhD. “We owe it to every participant and the public at large to support the maximal use of this knowledge for the greatest benefit to human health. This important commitment from researchers to research participants must always be upheld.”
Developed by NIH in close coordination with the FDA, the proposed rule details procedures for meeting the requirements established by FDAAA to improve public access to clinical trial information. FDAAA and the proposed rule apply to certain interventional studies of drugs, biological products, and devices that are regulated by the FDA, but, generally, not to Phase I trials of drugs and biological products and small feasibility studies of devices. The proposed rule specifies how data collected and analysed in a clinical trial would be required to be submitted to ClinicalTrials.gov. It would not affect requirements for the design or conduct of clinical trials or for the data that must be collected during clinical trials.
“This proposed rule would close an important gap, making additional information about clinical studies of investigational drugs, medical devices and biological products available to the public,” said FDA Commissioner Margaret A. Hamburg, MD. “It would help eliminate unnecessary duplicative trials, advance biomedical innovation, and provide the public with a much richer understanding about the clinical trials for these products.”
Notable changes from current requirements and practice that are outlined in the proposed rule include
• A streamlined approach for determining which trials are subject to the proposed regulations and who is responsible for submitting required information.
• Expansion of the set of trials subject to summary results reporting to include trials of unapproved products.
• Additional data elements that must be provided at the time of registration (not later than 21 days after enrolling the first participant) and results submission (generally not later than 12 months after completion).
• Clarified procedures for delaying results submission when studying an unapproved, unlicensed, or uncleared product or a new use of a previously approved, licensed, or cleared product and for requesting extensions to the results submission deadline for good cause.
• More rapid updating of several data elements to help ensure that users of ClinicalTrials.gov have access to accurate, up-to-date information about important aspects of a clinical trial.
• Procedures for timely corrections to any errors discovered by the responsible party or by the Agency as it processes submissions prior to posting.
Read a summary of the proposed changes: www.nih.gov/news/health/nov2014/od-19_summary.htm
Related News
-

News The next 15 drugs up for negotiation with Medicare include several blockbusters
By now, everyone is quite familiar with the drug price negotiations taking place between drug companies and the Centres for Medicare & Medicaid Services (CMS) in the USA as part of measures being taken to reduce the cost of drugs for patients, to make ... -

News PSCI Welcomes Delpharm, Samsung Biologics, and Suven as First Supplier Partners
The pharmaceutical industry continues to evolve with an increasing focus on responsible sourcing, sustainability, and collaboration across the supply chain. Under a new model to recognise suppliers within the pharmaceutical and healthcare industry that... -

News Drug prices agreed upon as part of the US Inflation Reduction Act
The Inflation Reduction Act brought into constitution by the Biden administation in 2022, which proposed a drug price negotiation between the government and pharmaceutical companies, has reached it's first agreement. -
.png)
News Eisai Alzheimer’s drug authorised in UK but still faces obstacles
In partnership with BioArctic AB, pharmaceutical company Eisai has been granted Marketing Authorisation by the Medicines and Healthcare products Regulatory Agency (MHRA) for its Alzheimer’s disease drug product Leqembi. -
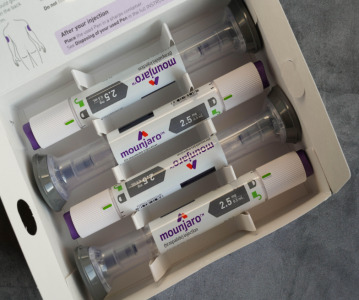
News Eli Lilly's weight loss drugs removed from the FDA's shortage list
The US FDA have recently updated their drug shortage list. The recently released list shows that all dosage forms of Eli Lilly's weight-loss drug Zepbound and their diabetes drug Mounjaro are now available. -

News Global advancements in the diagnosis and treatment of rare diseases: Rare Disease Day 2024
Rare Diseases Day is celebrated on the 29th February 2024 and represents the plight of rare disease patients to gain diagnosis and access to suitable treatment. -

News Pharmaceutical industry supports COP28 health stance in joint statement
As COP28 takes place over this week in Dubai, UAE, several bodies in the pharmaceutical and health industries have come together to announce support of key movements in sustainability in the sector, and to recognise sustainability as a health issue.&nb... -

News Biden backs Cold-War measures to shore-up medical supply chains
In a recent strategy to combat rising inflation and the cost of living crisis, President Joe Biden has invoked a Cold War-era act to increase investment in a selection of medicines and supplies.
Position your company at the heart of the global Pharma industry with a CPHI Online membership
-
Your products and solutions visible to thousands of visitors within the largest Pharma marketplace
-
Generate high-quality, engaged leads for your business, all year round
-
Promote your business as the industry’s thought-leader by hosting your reports, brochures and videos within your profile
-
Your company’s profile boosted at all participating CPHI events
-
An easy-to-use platform with a detailed dashboard showing your leads and performance